Single-cell RNA sequencing (scRNA-seq) has changed the way researchers study cells by allowing them to examine gene expression at the level of individual cells. This technology helps uncover subtle differences between cell types, identify rare cells, and track how cells change over time. However, the growing scale of scRNA-seq datasets makes it increasingly difficult to accurately and efficiently label or annotate these cell types, especially when researchers are working with complex tissues like the retina.
To solve this problem, researchers at Baylor College of Medicine have developed an accessible and highly accurate workflow using scGPT, or single-cell Generative Pretrained Transformer. scGPT is a large language model adapted for biological data. Instead of predicting words, it predicts patterns in gene expression. This transformer-based architecture, the same kind that powers tools like ChatGPT, has been trained to understand how genes behave across millions of cells. Researchers can then fine-tune the model on their own data to boost accuracy in classifying cell types.
An overview of the end-to-end workflow to fine-tune scGPT classifiers in large-scale RNA-seq datasets
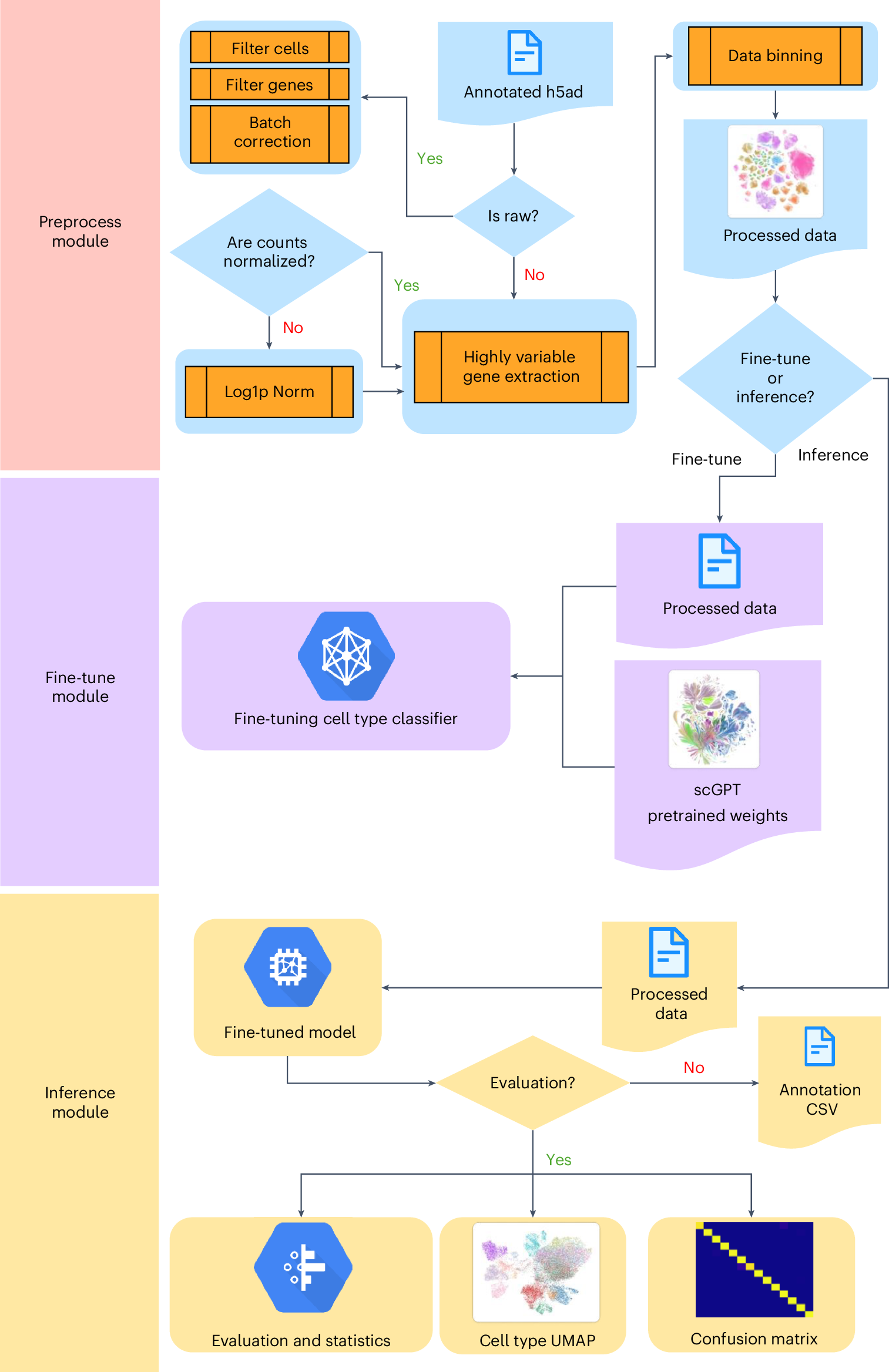
This figure outlines the detailed workflow in the Protocol. Before starting any task, whether fine-tuning or inference/evaluation, data preprocessing is always required. This step prepares the dataset by cleaning, normalizing, binning and compressing it into a new data file, which is then ready for use in subsequent tasks. In the fine-tuning module, the preprocessed data and a pretrained scGPT model are used to set up the pipeline, with the goal of further refining the model to learn more specialized data based on prior knowledge. The inference module, on the other hand, requires a fine-tuned model, which can either be a custom fine-tuned model or the pretrained scGPT model for zero-shot evaluation. At the end of the inference pipeline, key outputs include a UMAP for cell-type clustering and a CSV file with prediction results. If the inference dataset contains actual cell types, an optional confusion matrix will also be generated. The data can be accessed on Zenodo, and the code is available on GitHub.
In their protocol, the team focused on retinal cells as a demonstration case. Using a custom retina dataset, they trained scGPT and achieved a remarkable 99.5% F1-score, meaning the model was highly accurate in predicting cell identities. The workflow automates the most challenging steps, including data cleaning, training, and evaluation. Importantly, it is designed to be user-friendly even for those with limited coding experience. It includes both command-line tools and a Jupyter Notebook that guide researchers through the process from start to finish.
This tool isn’t just for computational biologists. With minimal knowledge of Python and Linux, researchers in many fields can now take advantage of high-performance AI tools to make sense of their scRNA-seq data. The protocol is adaptable to different tissues and datasets, making it useful for projects in neuroscience, immunology, cancer, and more.
By lowering the barrier to entry and providing high-precision annotations, scGPT enables researchers to unlock deeper insights into cellular diversity. As scRNA-seq becomes more central to biomedical discovery, tools like this will be essential for turning complex data into meaningful biological understanding.
Availability – The code for this protocol is available via GitHub at https://github.com/RCHENLAB/scGPT_fineTune_protocol.
